
Breath from Salt by Bijal Trivedi (Book Review)

As I sat through many of the workshops and plenary sessions at the 2023 North American Cystic Fibrosis Conference (NACFC) I couldn’t help but notice the upbeat energy of the conference. Having just read Bijal Travedia’s majestic history Breath from Salt about the history of cystic fibrosis (CF), it made all the more sense; we were in a new era of cystic fibrosis (CF). For most of the disease’s history, patients died as children, or maybe in their teens if they were lucky. Now, many individuals with CF are making it to retirement. Some estimates are even suggesting that children with CF born today may have close-to-normal life expectancy. For a disease with a life expectancy of 5 years in the 1950s, this change is absolutely staggering.[1]
Persons with CF (pwCF) and their family members are an extremely well organized group. While every developed country has a national CF foundation (e.g. Cystic Fibrosis Canada), the US-based Cystic Fibrosis Foundation (CFF) acts as the lodestar of the movement. As the first sentence of their mission statement makes clear: “The mission of the Cystic Fibrosis Foundation is to cure cystic fibrosis.” Amazingly, even though 95% of patients are now eligible for life-saving treatments, none of the momentum has been lost and CFF and broader research community is still pursuing a path to the cure. Success has emboldened the community to further success.[2]
The median age at death for pwCF has been rising dramatically
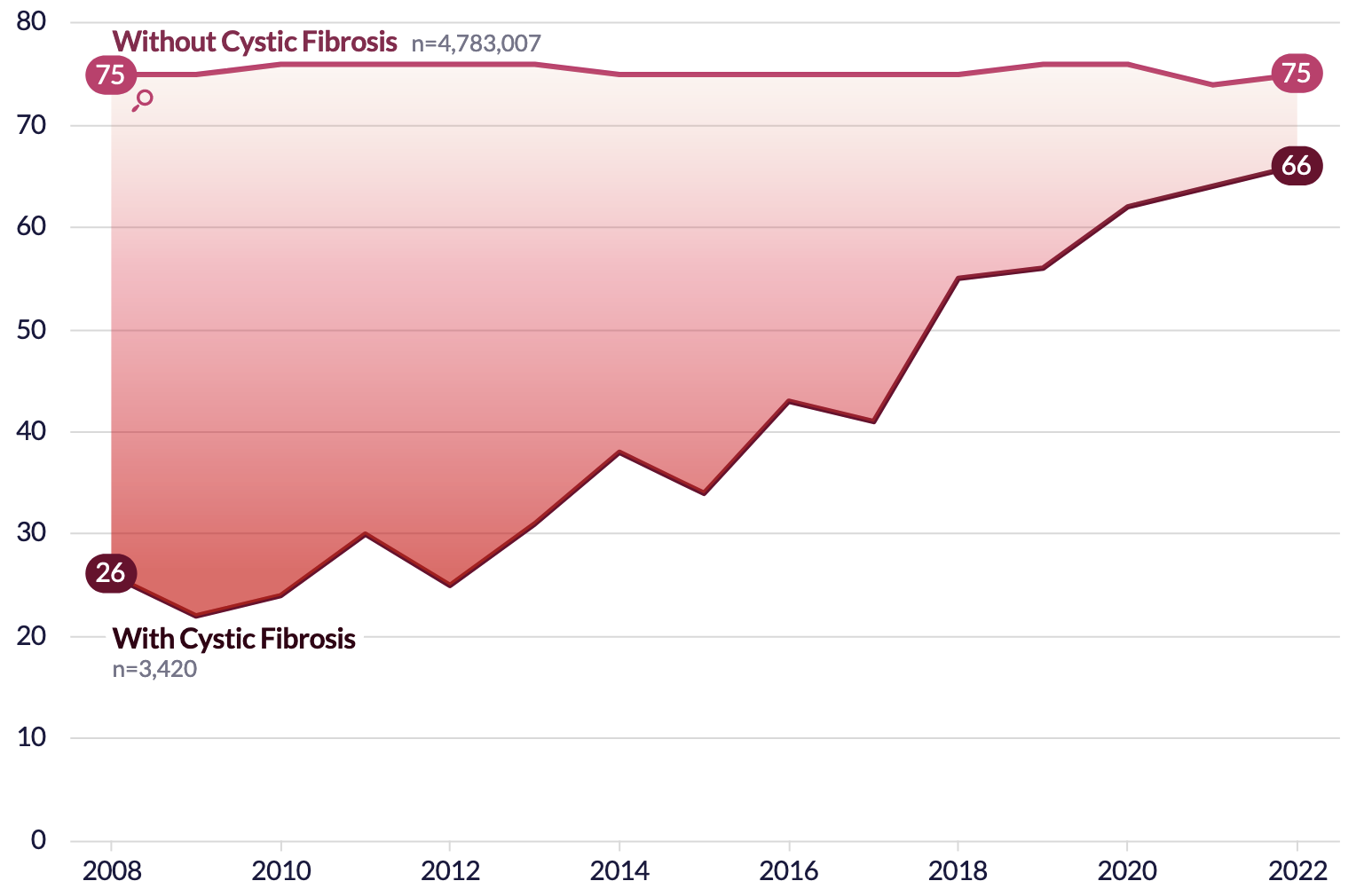
Source: Epic Research
More than 4500 individuals participated in the 2023 NACFC. There is now an army of countless researchers, students, postdocs, genetic counselors, nutritionists, physical therapists, nurses, doctors of all specialties, and companies who are engaged with this disease. At the poster session of the conference, I saw research that analyzed every aspect of the CF health ecosystem from optimal nutrition, to mental health, to cell line tests for ultra-rare forms of the disease that fall outside of the treatment perimeter. To put this in historical perspective, in the 1940s no one outside of maybe a dozen doctors even knew what the disease was.
To understand the amazing gains in CF, you need to understand both the remarkable history of scientific breakthroughs and the organizational structures that enabled them. Travedia’s new book provides an accessible and comprehensive history of the disease. I suspect this book will be the definitive account of CF until a new chapter of the disease is written with the next era of gene therapy treatments.[3]
Breath from Salt has strong parallels both in terms of quality and comprehensiveness with Emperor of All Maladies, Siddhartha Mukherjee’s book about the history of cancer. Like the other great writers of medicine, Travedia’s narrative blends history, politics, and science. Each discovery in the disease’s history is connected, and we can see how the accumulation of these discoveries led to the therapeutic breakthroughs of today. The book contains many powerful and tear-inducing stories of pwCF, and the impact that death has on families. Readers who are interested in organizational structures and research politics will also find this book essential reading to understand the pioneering venture philanthropy model the CFF used to fund and generate research for this disease.
For those who have never heard of CF or only have a vague impression of the disease, it is one of the most common rare disease in the Caucasian population that is also lethal.[4] CF is an autosomal recessive monogenic disorder, meaning if both pairs of your chromosomes have a mutation (recall you have a two pairs of each chromosome, one from your mother and one from your father), you will (almost surely) develop the disease.[5] The CFTR gene contains the instructions for making the CFTR protein. This protein facilitates chloride ions to move in and out of cells. These chloride ions play a key role in controlling how salty or acidic our body fluids and tissues are (recall that table salt is about 60% sodium and 40% chloride). In our bodies, the balance of these ions is crucial for normal function.
When this gene is not working properly, cells are unable to facilitate chloride ion transfer which results in dry and sticky mucus which accumulates in the lungs and prevents the pancreas from producing proper enzymes.[6] Over time, pwCF can experience chronic infections and a build up of bacterial colonies which can cause permanent damage to the lungs and other organs.[7] Historically, 1 in 6 of patients waiting for lung transplants had CF. There are 105K pwCF globally, of which the US (40K) and Europe (55K) make up >90% of the diagnosed population. Although CF is certainly under-diagnosed in places like China and India, the Caucasian population is at the highest risk for the disease.[8]
It bears repeating that the story of CF is not just fascinating because of the remarkable increase in life expectancy, but also because of the unique way that the CFF was organized and was able to push and incentivize treatments for CF. Similar to the way that the 18th century abolitionists created the modern concept of societal campaigns with pamphlets, chapters, and political lobbying, the CFF showed how the not-for-profit sector could push and incentivize pharmaceutical companies and researchers to study the disease they wanted and develop treatments by providing hundreds of millions of dollars of funding over the years. The CFF developed the idea of venture philanthropy, which has been emulated since by groups like the Gates Foundation, the Chan Zuckerberg Initiative, and other rare disease groups (like ALS and multiple myeloma). If you want to understand how a group of patients suffering from a rare disease can help to shape the funding priorities of granting agencies like the NIH, the topics researchers focus on, the product portfolio of pharmaceutical companies, and the donations from wealthy philanthropists, then the success of CF is the model to understand. Full stop.
The story of CF is still being written. But here is its remarkable journey so far.
Dum spiro spero
In the year 2012 the FDA approved the drug Kalydeco as the first pharmaceutical intervention for CF which actually targeted the molecular nature of the CFTR protein deficiency in those with the disease. How did this drug come to fruition? Normally pharmaceutical companies invest their own money (billions of dollars) into each drug that goes through clinical trials (most fail), but are able to stay profitable by (more than) recovering their fixed and variable costs by charging as much as the market will bear for as long as their drug has patent protection in the United States.[9] But Kalydeco’s development model was different from any other drug at the time. Though it was developed by Vertex Pharmaceuticals, almost all of the early exploratory work and costs were paid for by the non-profit CFF. Where did a non-profit get the hundreds of millions of dollars needed to align the incentives of commercial entities with CF patients? We’ll get to that later, but a businessman named Joe O’Donnell who spearheaded the fundraising efforts is owed a great deal of credit.
Cystic Fibrosis must been with our human ancestors for thousands of years (the F508del mutation likely emerged in the Bronze Age). But until the modern era, most children died before the age of 5, so it would have merely been another unexplained malady. In the 1930s Dorothy Hanson Andersen began investigating the curious circumstances around some children who died in New York City’s Babies Hospital. Babies born with CF who are pancreatic insufficient will be unable to produce the enzymes necessary to breakdown food and absorb nutrients and calories. Despite non-stop feeding they will fail to gain weight and will show symptoms of starvation like kwashiorkor (swollen belly). Babies who died in these conditions were labelled as having “celiac disease,” a catch all concept for those who were unable to absorb or take in food. But Andersen wasn’t satisfied with this overly broad explanation. In 1936, Andersen was performing an autopsy on a 3-year old girl known as MD and she noticed that while almost all of her organs were fine, the pancreas “was filled with fibrous cysts” and the pancreatic duct was “lost in a mass of tough scar tissue.”
It was clear that this organ couldn’t possibly have been doing its job. That explained the patient’s malnutrition. But Andersen had never seen a celiac patient with a pancreas that was so damaged. She began combing the medical journals for clues, spending all of her free time hunting through stacks of books and journals in nearby Columbia University’s library for mentions of similar cases, and exploring autopsy files of other children who had been diagnosed with celiac disease. She soon discovered reports from Boston to Europe to Australia describing similar fibrous pancreatic cysts in other children who had been classified as celiac patients. But she knew that kids with celiac disease, once they were prescribed the right diet, didn’t usually die; they grew quite normally. Perhaps MD didn’t have celiac disease at all. In fact, based on the state of the cystic, fibrotic pancreas, she was fairly sure she didn’t. Surrounded by an ocean of medical literature in Columbia’s library, Andersen realized this could be an entirely new disease - only that, unlike celiac, was incredibly deadly.
Andersen published the first academic paper describing CF in 1938, and from that point became one of the leading scholars and practitioners in the field, helping to pioneer nutritional regimens which extended the lifespan of children by many years. Until the end of her life in 1963, she would regularly correspond with parents from across the United States and was described as an “extra parent” to the children suffering from CF.
While no one knew the cause of this disease, thanks to Andersen’s work it was at least possibly to accurately diagnose children with it. Now physicians could give them the nutritional support they needed – a small but critical step toward keeping these children in good health for as long as possible.
The fog begins to lift
An old medieval saying reveals that our pre-modern ancestors were aware of CF: Woe is the child who tastes salty from a kiss on the brow, for he is cursed, and soon must die. The first written reference of CF has been found in 1595 by Pieter Pauw - a Professor of Botany and Anatomy at the University of Leiden - who noted that an 11 year old girl had a swollen hardened gleaming white pancreas. At the time, children born with sweaty skin were assumed to be “bewitched.”
In the late 1940s, a physician named Paul di Sant’Agnese was the first to connect the now well-documented pathologies of CF (pancreatic insufficiency and lung infection) with a seemingly more benign phenotype - that of salty skin. Andersen had hired Sant’Agnese to work at The Babies Hospital to accommodate the growing number of CF children that were coming every year. In the swealtering hot summers of 1948 and 1949, di Sant’Agnese noted something strange: many of his patients were showing up with heat stroke - at one time all of children admitted. A further curiosity presented itself:
Sitting in a room with one patient, di Sant’Agnese notice that when the little boy put down his glass after gulping down water, the surface was decorated with ghostly white, salty fingerprints. The other children left similar prints, which inspired the physician to begin investigating the physiology of sweat.
Sensing the potential for a diagnostic breakthrough, di Sant’Agnese ran experiments on 43 individuals with CF and 50 without, and found that the chloride levels in the CF population was 3-5 times higher than those without the disease. The concentrations of chloride that di Sant’Agnese found in the early 1950s (60-160 mEq/L), represents the current threshold used to diagnose CF patients (>60 mEq/L). Not only did these findings point to a radical and somewhat baffling molecular origin of CF, on a more practical level they meant that CF patients could now be diagnosed with a sweat test rather than a highly invasive “duodenal procedure” which tested for enzymes found in the small intestine.[10] By 1959 the physicians Gibson and Cooke and had refined the sweat test method which is almost identical to the diagnostic test used today.
Based on the history outlined in Breath from Salt, I would assert that the modern CF era began in the 1950s. This decade saw the creation of reliable diagnostic testing, the first reference of CF in a medical textbook (penned by di Sant’Agnese of course), and the first national organization in the US that started to raise awareness about the disease and began collecting funds for research that might one day lead to treatments. In 1948 when di Sant’Agnese first noticed the “salty fingerprints,” there was no treatments for the disease beyond nutritional regimens, there was no support for parents beyond a handful of experts like Andersen, no one knew what caused CF, and no one seemed to be looking into how the disease might be actually be treated. Every new diagnosis of CF at that time was therefore a tragedy akin to a childhood cancer. But with each draw of this horrible genetic lottery, a growing population of grieving families began to demand change and answers for their children.
In 1953 the first son of a wealthy socialite named Wynne Sharples was born with CF, and a year later their first daughter was as well. Wynne’s family was sufficiently well-connected that her father was able to set up a meeting of the director of Boston Children’s Hospital who in turn summoned Harry Shwachman, one the leading CF physicians, who agreed to personally supervise the care of her children. Wynne knew about some of the other grassroots movements that had started up, but was uninterested in participating in efforts she considered to be too amateur. She understood that advancing research on this disease would require bringing together top scientists to find a cure, wealthy society types to bring awareness, and funds, and politicians who could wield influence. In her vision, Wynne saw local grassroots as feeders which could fund a higher profile national foundation.
The inaugural meeting of the National Cystic Fibrosis Research Foundation (NCFRF) was held in 1955 in Philadelphia. Early on it was clear that there was going to be tension between the vision of the national foundation, and the grassroots organizations being run by parents who wanted any help they could get treating their increasingly sick children. This initial tension would continue for the next 50-60 years until most pwCF had access to modulator therapies. But at a time when not only was there no pharmaceutical treatment on the horizon, and the underlying cause of the disease was unknown, the prospect of spending money on basic research while families desperately needed help with nutritional supplementation, constant hospital visits, and physiotherapy must have been a wrenching decision.
As president of the NCFRF, Sharples’ top priorities in 1957 were education and finding a research director. The foundation put together booklets and quarterly newsletters for foundation members and Sharples recruited Andersen, di Sant’Agnese, and Shwachman to review the material.
The research director would be a foundation employee tasked with knowing everything possible about the disease, including which researchers would be most likely to study it. That same individual would also be in charge of encouraging those researchers’ interest in this rare condition. Money, Sharples knew, was the only way to fund research and develop a cure. She hoped that as the public learned more about the disease they would be generous and provide the funds.
In order for the public to learn more, the NCFRF ensured that newspapers across North America mentioned the disease. In the Bergen Evening Record from April 12th 1957, Sharples made clear that,
… “Our mothers and fathers, together with their friends, are encouraged by the example of the polio foundation. It was with the persistence of local chapters which raised the necessary funds to mount the all-out assault on polio, leading to a successful vaccine. Intensive medical research alone will enable us to solve the problem of cystic fibrosis. We know that we can count on the generous support of American parents toward this end.”
Later that year, a NYT article declared a “War of Cystic Fibrosis,” and also made references to the success that the National Foundation for Infantile Paralysis (NFIP) - aka the “March of the Dimes” - had for the development of a vaccine for polio. The timing for CF was fortuitous. The astounding results of Salk’s polio vaccine were released in 1955, and the news of the vaccine’s effectiveness was greeted with an outpouring of public joy. Church bells rang, factory whistles blew, and spontaneous gatherings occurred in streets and public places. People were overjoyed and relieved, as the vaccine promised to end a terrifying health scourge. Importantly for CF, this seminal biomedical event helped to shape the public perception that publicly funded scientific advances could cure diseases. Before the vaccine, thousands of children were paralyzed every year, afterwards, almost none were.
Doris Tulcin was another of the key founders of the NCFRF and leveraged her family’s connections to run high-profile fundraisers, as well as increase the ties between the NCFRF and Basil O’Connor, the president of the NFIP. Although O’Conner suggested to Doris Tulcin and George Frankel (Doris’ father) that they consider merging the NCFRF with the NFIP, the idea was politely rebuffed. But, with agreement from the NFIP, George suggested that they recruit seasoned staff from the NFIP to join NCFRF to replicate some of the magic they achieved with polio for CF.
Basil distilled for Tulcin and her organization the key lessons he lad learned from polio and the NFIP.[11] The first was that if the public was going to help fund raise for the disease they needed to know about it and feel connected to the cause. The NFIP used public relations experts to gain national exposure, and came up with innovative national campaigns like the “March of the Dimes.” The second was that fundraising would be key to generating the funds needed for research. Money was money, and the NFIP used everything from high-brow black tie events to local neighborhood fundraisers like bake sales to national campaigns. Third, and most crucially, funds should be earmarked to research rather than patient care. While helping patients is more compassionate in the short term, the longer term gains from a treatment will always outweigh any short-term humanitarian impulses. The NFIP originally prioritized helping patients through services like the Warm Springs Institute for Rehabilitation, but later changed tack.
Hired in 1946, Harry Weaver was the NFIP’s director of research who took a “ruthless approach to research funding… [and] single-handedly catalyzed the pace of polio research.” The NFIP designed short-term grants that were designed to answer specific questions that would move the science closer to a vaccine. If the research showed promising results, the grant was renewed, if not, it was terminated. Weaver also supported funding competing approaches to vaccines, believing that the natural competition between scientists would catalyze their work (especially when their pet theories were on the line). But how to get actual scientists to apply for grants and take an interest in the disease? Weaver stressed the importance of cultivating goodwill with the scientific community and ingratiating oneself with the universities by paying for new facilities and their overhead expenses. After a few years of going all in on research, the science of polio had progressed to the point where a full scale vaccine trial could be run. The NFIP spent $55 million to run a trial of more than 1.3 million children in 1954 (which still constitutes the largest clinical trial even run), and made sure that no cost was spared. All ancillary trial costs were covered including the subsequent statistical analyses. When the rollout of a life-preserving vaccine is at stake, it’s foolish to be miserly when a single day can be the difference between health and sickness.[12]
Every one of these recommendations would in one way or the other be leveraged by the NCFRF (which became the CFF) and the CF community more broadly. While CF could leverage these insights, the situation was quite different compared to polio. Only a small number of children were at risk of being born with CF every year (in contrast every parent was afraid their children might develop polio), polio had the President of the United States (Franklin D Roosevelt) as a brand ambassador, and the existing science of inoculation (while still young) would prove sufficient for finding a cure. Not only would a vaccine not be possible for CF, no one even knew what caused the disease or what a treatment would look like.
Show me the data
Less than five years after its launch, the foundation had gathered significant strength, power, and visibility, and they were ready to launch what they saw as the next stage in the battle against the disease: the creation of centers dedicated to treating and caring for children with cystic fibrosis.
Due to disagreements with the leadership at the NCFRF, including with Dorothy Anderson, Wynne Sharples agreed to step down and a trucking executive named Robert Natal assumed the role in 1960. These were critical years. Kenneth Landauer had become the foundation’s new director of research and education and helped to found a series of medical centers for cystic fibrosis patients across the country. The idea was not new, and Landauer drew on his experience having set up similar centers for polio. However, the need for these centers proved to be short-lived after to the rapid success of the polio vaccine. However, cystic fibrosis wasn’t disappearing anytime soon, and CF patients desperately needed standardized protocols and treatment practices that were grounded in evidence-based medicine. The NCFRF was essential in ensuring that any centers in this network actually had an incentive to adopt best practices and maximize patient outcomes.
But what was the data that could be used to determine the best practices for the treatment of CF? And furthermore, what exactly was CF? True, Dorothy Anderson had shown fibrotic cysts found on the pancreas distinguished CF kids from other disease types, but the variations in disease progression and symptoms varied substantially. Only by concentrating pwCF into care centers could the range of clinical symptoms be documented, and crucially, form the basis of knowledge for practitioners in this space.
Based on the polio experience, these initial CF care centers were focused on both treatment and research, and could apply for unlimited funding from the national foundation for the latter. These two policies, creating academic medical centers with access to deep research funds led to a mini-boom in the number of PhDs studying CF. The advice O’Conner had passed on to Tulcin and that was pushed by Sharples was now being executed by Landauer. Institutions and money were creating a CF research agenda.
Anyone who had analyzed healthcare data or studied healthcare economics knows a fascinating, and arguably disturbing, phenomena: the quality and cost of medicine varies tremendously across providers. Atul Gawande famously referred to this as the bell curve of medicine. We all understand that there is going to be variation in medicine. Sometimes a hospital is short-staffed, a doctor is a recovering from a flu, an X-ray technician hasn’t slept in 24 hours, etc. But the statistical estimates of the variation are often of a concerning size. By 1964 it was clear that one CF care center, the Babies and Children’s Hospital in Cleveland, was doing better than any other CF center, and by a long shot. Most children with CF were dying at the age of 3, but somehow in Cleveland they were dying at the age of 21. How could life expectancy be 7-fold higher?
In 1955 Cleveland established its own local branch of the NCFRF and shortly afterwards convinced the chairman of pediatrics at the Babies and Children’s hospital to launch a research-based treatment program for CF. A 29-year old doctor named LeRoy Matthews was tapped to the run the program. While Matthews found the prospect daunting, he was a man who never lacked self-confidence, and saw the tremendous opportunity this represented for a young physician. Matthews was Harvard educated and had previously studied under Harry Shwachman and Sidney Farber, giving him an amazing set of connections and knowledge about a disease that was largely unknown in the developed world at the time.
He approached the problem analytically and did a whistle stop tour to centers with existing CF practices. In Boston, Shwachman told Matthews about his physiotherapy regimes that percussed the chest with vibrations so that the mucus could be coughed up. He also learned about Shwachman’s low-fat nutrition regimen and shock and awe multi-course antibiotic treatments to “pummel the tenacious bacteria infecting the lungs.” At the Babies Hospital in New York, Matthews learned from Anderson and di Sant’Agnese about different diagnostic testing strategies for the disease as well as its nuances that only decades of practice could afford. Moving onto Philadelphia, Wynne Sharples and her husband Dr. Robert Denton, who was a pediatrician, showed Matthews his nebulizer invention that helped to loosen the hard mucus.
Within a few years of opening his clinic, Matthews had cut the mortality rate substantially. His analytical approach was matched with an equally impressive ability to connect on a personal and emotional level to both patients and parents. Constant vigilance is what determined the difference between mediocre and exceptional outcomes in CF. The requirements around percussive physiotherapy, oral antibiotics, and nebulizers were strict and challenging, but could also provided a sense of accomplishment and participation. While Matthews was admired for his dedication by his patients, his colleagues were not as impressed, especially as he enjoyed touting his success loudly. He once told one conference of physicians that “how long [our patients] will live remains to be seen, but I expect most of them to come to my funeral.” His braggadocio was met with skepticism and jealously by other physicians, many of who assumed Matthews’ data was simply fake.
In part to settle the dispute between Matthews and his skeptical counterparts, a young pediatrician named Warren Warwick (who would later co-invent the first mechanical vest to help clear the airways of mucus) created a one-page questionnaire for each CF center to complete for each patient at their center. Warwick was meticulous and his small team ensured that the data was complete across the 30 centers that existed at the time and was anonymized to ensure analysis would not be biased. Sure enough, the results in the early 1960s showed that Matthews’ clinic claims were true: his center had a rock-bottom mortality rate and average age of death of 21. The skeptics were not convinced and another analysis was done by Cecil Nesbitt who recapitulated the results.
Having unambiguously demonstrated that Cleveland’s CF clinic was doing something special, Matthews worked with Landauer to create national treatment guidelines based on his center’s approach. The survey that Warwick developed would now be used to create annual reports for each center, which would show the rank of that center in comparison to others. Warwick was asked by other CF foundations in Canada, Australia, England, and Sweden to create similar surveys and reports for them as well.
The patient registry that Warwick originally crated for the NCFRF would eventually be replicated by other disease nonprofits and biomedical institutions across the world. Though neither Warwick nor the foundation’s leaders could have known it at the time, the patient registry would eventually prove to be the foundation’s most valuable asset–key to understanding the progression and nuances of the disease–and, decades later, it would further revolutionize patient treatment.
The end of the beginning
By 1980 things had improved markedly for the CF community since Anderson’s paper was published nearly 40 years before. Many babies were receiving accurate diagnoses much earlier, and being treated with prophylactics that targeted the symptoms of the disease. Life expectancy was now in the mid to late teens. But the fundamental biology, or etiology, of the disease was still unknown. By the end the decade however, the fundamental biology of CF would be elucidated, giving the tantalizing hope that a “cure” would be around the corner.
Everyone knew that CF patients had salty sweat, with chloride levels showing variation but usually being above 60 mmol/L. But besides cases of extreme heat, this salty sweat didn’t in and of itself point to anything sinister. Yes, something about the genetics of the CF disease was giving rise to this phenotype, but how did salty skin connect with any of the other pathologies like lung infections or pancreatic insufficiency?
Dr. Richard Boucher was doing experiments in the late 1970s on the physiology of lung diseases. He knew that in diseases like asthma or COPD the mucus in the lungs was too dry. Normally our mucus is around 98% water, 1% salt, and 1% mucins. This balance between dry and wet is carefully calibrated so that the mucus is sticky enough to trap unwanted pathogens, but fluid enough that our cilia (broom-like hairs) can “sweep” the mucus up the lungs so that we can cough up the gunk that we incidentally inhale. Boucher hypothesized that since water is attracted to electrolytes (like chloride and sodium), perhaps these diseases are caused by disruptions in salt transfer across the cell membranes. Luckily for a researcher, whenever electrolytes move they generate electrical activity, meaning that voltage measurements could be used to compare differences with healthy volunteers.
In the case of asthma, Boucher’s hypothesis was wrong, the voltage was the same for healthy and asthmatic patients. But Boucher knew of CF and the associated sweat test. Now at UNC, Boucher teamed up with another young physician Michael Knowles where they tested the voltage of the nasal passage between normal and CF patients. The results were astonishing: CF patients had voltage levels three times more (negative) than healthy controls. While nasal cells are a good proxy for lung cells,[13] Boucher and Knowles needed to be sure the results were replicable in the lung cells of CF patients where the actual problems occurred.
On the verge of a massive breakthrough Boucher and Knowles needed to confirm whether the problem of “sweaty” cells was caused by chloride, sodium, or both. First they applied a medication which would block sodium being able to move into the nasal epithelial cells. Sure enough, the voltage reading was now back at the normal level. Next, they rinsed the nose which temporarily removed the chloride. In non-CF patients the chloride returned the surface of the cell which could be registered as a large negative voltage. However, for the CF patients the voltage remained unchanged meaning that the chloride was not getting replaced at the cell surface. Boucher and Knowles had now demonstrated at the chemical level what was going on: too much sodium flowing into the cells, and too little chloride flowing out, both of which caused the mucus to become drier. In other words, with chloride being unable to leave the cell, the cell’s natural mechanisms which maintain the electrochemical gradient and fluid balance are activated and sodium channels become more active, drawing the sodium and therefore water into the cell.
But as revelatory as this new information was, the implications for treatment were unclear. Besides, cystic fibrosis didn’t just affect the lungs, it damaged multiple organs and affected the sweat glands, too. The work was clearly far from done.
Around the same time and Boucher and Knowles, Paul Quinton was also looking for a root cause of CF, although his quest was personal. Quinton had grown up in rural Texas and had diagnosed himself as a teenager. While doing his PhD at Rice, Quinton experimented with his own sweat on frog cells, although initially promising results were shown to be flukes. Undeterred, Quinton continued his research by doing a postdoc at UCLA. Once again, Quinton used his own body as the material for laboratory experiments. Aware of voltage results from Boucher and Knowles experiments, Quinton replicated the findings with microscopic sweat ducts he had extracted from his own skin cells and healthy controls. Quinton’s further experiments confirmed that while sodium was able to pass in and out of his sweat ducts, chloride wasn’t, suggesting that “this chloride transport defect may be closely associated with the fundamental disturbance in this disease.”
Chloride, everyone now agreed, was the electrolyte causing problems in the nose, lung, sweat glands, and possibly the other organs as well.
In 1974 Bob Beall (pronounced “bell”) had joined the National Institute of Health (NIH) in Bethesda, Maryland. In the US, the NIH is largest funder of medical research, around 20% of the total.[14] Beall was attracted by the public policy focus of the NIH and became the director of their Metabolic Disease Program, which at the time was focused on the skyrocketing rates of diabetes. In 1976 Beall reluctantly agreed to a scientific meeting about CF (which he had never heard about). Beall was won over by the passion of the small and desperate community who was only able to present a paltry amount of evidence about the disease at the time. But even if Beall wanted to help, nothing could be done until the US Congress directed the NIH to study CF and earmarked the appropriate funds. That same year, the NCFRF changed its name to the now-known CFF and launched a lobbying campaign. It was successful. Congress ordered the NIH to conduct a study to look into the future research direction of the disease.
Over 1977-78 Beall, Tulcin, and the then-president Robert Dresing worked with Beall and other NIH scientists to put together a report Cystic fibrosis: state of the art and directions for future research efforts. From a handful of grieving parents raising money through bake sales in the 1950s, within 25 years the CFF had become a national organization with the imprimatur of the NIH and Congress. Researchers across the country now would be able to apply for funds to do CF research,
… [b]ut grants were for young novice researchers and too small to lure older, preeminent scientists with broad visions. Tulcin and Dresing aimed to build a critical mass of CF science, not a smattering of research here and there. They wanted a network of multidisciplinary centers of excellence, each with a couple of dozen researchers investigating different elements of this complex disease, all sharing data and collaborating. To make this vision a reality and have a shot at curing it would take substantially more funding than Congress would give and more resources than the NIH was willing to devote. If Dresing and Tulcin were going to create this research network… they needed Bob Beall to launch and lead it.
The CFF voted to once again move their headquarters, this time to Bethesda, Maryland where they would be right beside the NIH, and a stones throw away from DC. Beall was poached, and became the foundation’s director of medicine and science in 1980. He would leave the CFF 35 years later, having been the president for 21 of those years. Beall and company would plan “a whole new era in CF research–with dozens of research centers devoted to finding a cure.” While the handful of researchers at each CF center were doing great work to understand CF and its treatments better, Beall was convinced that only a “research agenda equivalent in scope to the one that Harry Weaver had orchestrated for polio” would be able to find a cure. And scope meant money.
Tulcin and Dresing launched a massive fundraising campaign which enabled Beall to approach universities with $500K grants which at the time was a sizable sum. Over 1981-83 three CF research centers were set up at the University of Alabama, UNC Chapel Hill, and UCSF. By 1994 a total of nine were set up in the US, and over 100 full-time researchers were dedicated to doing basic and applied research that would be aimed at developing treatments for all aspects of CF including the lungs, pancreas, and digestive tract.
Under the hood
Everyone knew that CF was an autosomal recessive disease, but no one actually knew what gene caused the disease. In 1983, Huntington’s disease was found to be on Chromosome 4, making it the first gene to be successfully “mapped.” However, gene mapping here merely refers to finding a genetic variant that is highly correlated with the actual disease-causing mutation. This phenomena is known as linkage disequilibrium in genetics, since pairs of co-occurring mutations (that might have arisen in evolutionary history) will be inherited together with a probability inversely proportional to their distance on the same chromosome.[15]
Lap-Chee Tsui (pronounced “choy”) was a geneticist who had come to the Hospital for Sick Children (SickKids) in Toronto in 1981 to join a dedicated CF research team. Like other researchers at the time, Tsui’s team was trying to use genetic markers to discover the location of the CF-causing gene by comparing the genetics of patients with and without CF. A seemingly mutually beneficial collaboration with a private sector company called Collaborative Research, Inc. was started in 1984. Collaborative had access to a much larger library of genetic markers that could be used to find CF, and the commercial prospects for testing for CF were immense.[16]
However, the academic/commercial relationship quickly soured when Tsui realized that Collaborative was holding back its results for fear of letting competitors get a lead. Using Collaborative’s markers, Tsui’s team discovered that the gene almost certainly was on Chromosome 7. However Collaborative tried to stall the results and Tsui was unable to disclose the discovery. Nevertheless, rumors started spreading that the CF-causing gene was on Chromosome 7 causing other labs to pivot. In 1985, Tsui was finally able to publish his results, which had been replicated that same year by two other researchers.[17] Everyone knew the chromosome, but now the gene itself had to be found. This was no small feat given that Chromosome 7 has roughly 159 million base pairs. Randomly selecting a location in the genome would be equivalent to randomly selecting a letter in a book that is roughly 60 times the length of the King James Bible.
Two years latter, Dr Williamson’s group claimed to have found the gene, but the result was soon proven to be erroneous, and instead the group had merely found another marker. Although the (false) result temporarily sent demoralizing shock waves through the labs hunting for the CF gene, the race was quickly revived. What would become the most famous collaboration in the history of CF research began in 1987 when Tsui and Francis Collins met at a genetics conference and agreed to join forces in their hunt for the gene.
The first solid piece of evidence for the gene’s location came at 6pm on May 9th, 1989 when an undergraduate student rushed into Tsui’s office and showed him that one CF patient was missing three base pairs, TTC, compared to a healthy patient. This was the F508del mutation. A futher analysis of 50 CF patients showed that 70% of them were also missing this triplet. But Tsui was cautious by nature, and his experience with the Williamson debacle reinforced his view that they needed to have rock-solid evidence before popping the champagne. First, they checked that the protein associated with this mutation had a shape that could plausibly be related to CF. The team’s biochemist said indeed it could be a transporter, aligning the protein’s form with the findings of Quentin and Boucher. Next they checked the DNA patterns of the parents, and it indeed appeared the F508del mutation followed the exact pattern of an inherited autosomal recessive disease. Lastly, a statistical analysis was run showing that this mutation almost certainly had to be the cause of the disease. Tsui was satisfied and christened the gene the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR).
The results sent shock waves through the research community and national media. The result was a triumph of North American collaboration, and (still) forms a foundation of pride for SickKids and UMichigan. The scientific work was split into several papers. Was a cure now around the corner? Collins and others tried to temper expectations, but a certain giddiness pervaded the community. Shortly after this discovery, CF treatments would reach their peak of inflated expectations, only to tumble during 1990s after failed gene therapy treatments. But that would be several years away.
Still, the finding was astounding. Cystic Fibrosis was the first disease to have its gene fully sequenced. But the initial discovery was already posing awkward questions. What about the 30% of other CF kids whose 508th amino acid was present? Why didn’t all kids have the same mutation. This finding launched one of the most important branches of CF research since 1989: chronicling the menagerie of variants which could lead to the disease. One of the Tsui team members, Batsheva Kerem, would go onto show within a few years that the majority of Ashkenazi CF patients actually had a different mutation called W128X. Unlike other mutations which led to a different or missing amino acid, this nonsense mutation led to a truncated (and effectively useless) protein. Although it couldn’t possibly be known at the time, these subtle differences in why the CFTR gene was malfunctioning would decades later determine which CF patients would and wouldn’t be eligible for treatments.
Glass half full
The 1990s was the first era in CF where multiple drug trials were conducted. The most dispiriting of these were the unsuccessful gene therapy trials that would leave the CF community in a funk. But the decade actually saw several important therapies including Pulmozyme and TOBI. Most critically, it was the decade where the CFF’s venture philanthropy began to pay dividends (pun intended). These few commercially successful products showed that i) the CFF’s investment strategy could work, and ii) Pharma companies could make a good rate of return on successful medications for CF. The latter detail was helped by the Orphan Drug Act of 1983, which was pushed for by a diverse coalition of interest groups including the CFF. This piece of legislation increased the financial returns for medications for rare diseases by increasing the length of patent protection, waiving various fees, allowing for additional tax deductions, and permitting expedited reviews.
Genentech was one of the pioneering biotechnology companies of the 20th century. They were the first to show how to mass produce insulin through recombinant DNA – basically getting bacterial cells to produce proteins. A bacterial plasmid is temporarily cut open, a new sequence of genetic material (usually a gene) is inserted, and then the plasmid is re-sealed. Billions of bacteria grow with this new gene with each organism producing a small amount of the inserted protein, allowing for production of a protein like insulin by simply giving bacteria sugar water. The “cutting” and “sealing” of bacterial plasmids is done by restriction enzymes and DNA ligase, respectively. This early genetic engineering allowed for the production of insulin and human growth hormone at a scale that revolutionized treatment of these diseases.
Steven Shak was a pulmonologist by training who had left the prospect of an academic career to join Genentech in 1986. Shak had treated CF patients before, and new that earlier research from 1961 had shown a “direct correlation … between the degradation of deoxyribonucleic acid and the decrease in viscosity of the tracheobronchial secretions from cystic fibrosis.” In other words, if you get rid of the DNA garbage (caused by all the exploded immune, bacteria, and lung cells), lung cells would become less sticky (viscous). Shak knew that DNase enzyme from cows would cut up DNA and theorized it might help to get rid of the DNA “gunk” in CF patients lungs. After spending many months finding the human (rather than bovine) version of the enzyme and synthesizing it, Shak contacted the Stanford CF center to obtain samples of sputum from pwCF.
When the enzyme was added the results were remarkable, and it wasn’t long before Tulcin and Beall came out to see a live demonstration. The CFF was all in, and promised to help with the clinical trial by creating the protocol, identifying the right patients, and engaging CF physicians, as long as Genentech would build the manufacturing capacity for producing DNase at scale.
The phase III results of the trial came out in 1993 and showed that pulmonary exacerbations (PEx) were reduced by 28%. Reducing PEx by a quarter was not world-shattering, but it was significant. The accumulation of PEx over the lifetime of CF patients was what ultimately led to lung deterioration. Furthermore Pulmozyme was arguably the first drug designed to fight a CF symptom, rather than a more general-purpose treatment like antibiotics.
In the early 90s, Bonnie Ramsey and Arnold Smith were two scientists at Seattle Children’s hospital working on developing a new antibiotic treatment for CF patients’ lungs. Based on anecdotes, they knew that some CF patients were inhaling their antibiotics, a potentially risky route of consumption given some of the chemicals in the medication. They wondered if they could develop an inhaled version of tobrmacyin that could be blasted deep into the lungs at high concentrations.
Aerosolized antibiotics conceptually made sense for CF patients, as their bacteria were hiding in the gunk and phlegm of their lungs. The CFF helped to support the research and the phase I and II trials which were shown to be safe and promising. Safety for inhaled tobramacyin was essential, as it could cause hearing loss and kidney damage at high concentrations when administrated intravenously.
Beall was able to find a company named PathoGenesis who was interested in commercializing the inhaled antibiotic. The company didn’t have much to lose, it was a startup and in need of its first win. The other Big Pharma companies were not interested in a route of administration that had never been approved by the FDA. But PathoGenesis knew that in addition to the Orphan drug status the product would receive, working with the CFF meant that almost any medication would be “self selling” since the entire prescriber base (CF physicians) would know about the treatment. Advertising is expensive, and removing this line item is a huge boon for any startup.
The company hired Bruce Montgomery from Genentech, the scientist who had developed the nebulizer for Pulmozyme. The company was able to solve many of the economic and logistical challenges including massively increasing the efficiency of delivery and stabilizing the formulation for shelf life. Before the phase III trial could start, Seattle Children’s and the CFF were at loggerheads about ownership of data and IP. PathoGenesis proposed a compromise:
[A] 5 percent royalty on sales if the drug went to market… The royalties would be fair payback for all the time, money, and effort both… had invested… This was the first time a health nonprofit had ever raised money through philanthropy and invested it in a for-profit company in return for future royalties.
Like Pulmozyme, the results for the now-named TOBI were impressive: lung function expanded by 11% and hospitalization declined by 25%. The FDA approved the treatment in 1997. The CFF would later sell the TOBI royalty rights to fund the therapies that have revolutionized CF treatments.
Trough of disillusionment
Within a few years of the CFTR gene being sequenced, multiple CF human gene therapy trials were poised to begin. The rate of progress must have felt dizzying. Within a year of the CFTR’s genetic code being released, two teams being led by Michael Welsh and James Wilson had used a vaccinia virus and a retrovirus, respectively, to get two different cell lines to produce the CFTR protein. The press coverage was instant and upbeat: “Cystic Fibrosis Corrected in Lab.”
While showing CFTR production ex vivo (i.e. outside of a living organism) is impressive, before a gene trial in humans would be considered, scientists would need proof they could resurrect the protein in a living animal model (in vivo). Gene therapies usually use viral vectors to deliver their payload (i.e. the genetically correct spelling of the gene), meaning the virus needs to infect the right cells (e.g. lung cells) for the effect to take hold. The first human gene therapy trial was started in 1990 for children with a genetic defect that left them immunodeficient. The results appeared successful, further fueling the hype and excitement around gene therapy.
At the end of 1992, both Welsh and Wilson were preparing to launch human trials for CF gene therapies along with Ronald Crystal. In 1993, Crystal’s team started phase I of their trial. Phase I clinical trials are designed to test the maximum dosage which is safe to administer by incrementally increasing the dosage amounts on relatively health patients. Crystal’s trial would test their gene therapy on the lungs of four CF patients. After the drug was applied to the third patient, something went wrong. The patient, a young woman, experienced a pneumonia-like condition as her oxygen levels crashed and her temperature and heart rate spiked. Further investigations revealed that the woman’s immune system was mounting an attack against the cells which received the viral payload. Although the trial was stopped, the investigators weren’t deterred:
An important lesson … is that despite extensive planning, animal studies, and thorough review, pre-clinical studies do not necessarily predict the response of humans (particularly individuals with disease) to gene therapy vectors. Of the animal models available, none mimic the human lung in CF. Thus, despite the lack of clinically evident toxicity observed in animal studies, human studies with viral vectors such as AdCFTR must be approached with caution. Despite this caveat, only human studies will permit the definition of the “efficacy-toxicity” window relevant to gene therapy. … [O]ur early data suggest that there may be a window in which Ad-mediated gene therapy could be used to treat the respiratory manifestations of CF.
Welsh’s trial differed in that they would only apply their gene therapy treatment to a small patch of nasal tissue, rather than the lungs. While the results from this approach would be less generalizable, they would be easier to implement and less risky. Their results were exactly what was hoped for: voltage of the nasal passage was restored to normal levels–a nice homage to work of Boucher and Knowles a decade earlier. The researchers were eager to publicize their results, and once again the media was more than willing to oblige, declaring a “[b]ig victory in war on cystic fibrosis.”
Welsh’s study was based on three volunteers. But as they expanded the number of patients, they noticed that the voltage change was no longer happening. Just like Crystal’s group, new patients were reacting differently. Wilson’s team, like Crystal, was also administering the therapy to the lungs, but in baboons rather than humans. Their toxicity study presented some reds flag, and the paper concluded that “adenovirus-mediated gene transfer into the lungs of baboons is associated with development of alveolar inflammation at high doses of virus.” To compound their problems, the effectiveness of the therapy was waning after a few weeks.
Beginning in 1995, the lackluster results from the human CF gene therapy trials in the lungs had planted a poisonous seed of doubt that such therapy may not be as simple as scientists had anticipated, and people started to lose faith. For his part, Wilson regretted that he hadn’t expressed his doubts more forcefully and managed expectations better, so that when the therapy failed, it would be less of a letdown. But the pressure to succeed was enormous, and everyone–the scientists, the patients, the foundation, the universities, and also the public–wanted gene therapy to work.
Scientists continued to experiment with different vectors and systems, with the goal of improving their safety. One therapy even got as far a phase II trial before concluding that “repeated doses of aerosolized tgAAVCF were safe and well tolerated, but did not result in significant improvement in lung function over time.”
The coup de grâce of the first era of gene therapy came in September 1999 when a young man named Jesse Gelsinger died after receiving gene therapy for a rare disease at UPenn. The fallout was a complete mess. The Gelsinger family sued Penn and Wilson (the lead physician), highlighting the conflict of interest the latter had with the biotech company Genovo that had the rights to commercialize the therapy. Subsequently, Penn, the FDA, and the Department of Justice all launched their own investigations, and the FDA halted dozens of trials in the subsequent years.[18]
By the early to mid-2000s, most researchers and companies would abandon efforts to use gene therapy to cure cystic fibrosis. The field was too immature and the tools and technology too crude. And for CF in particular, gene therapy no longer looked the best way forward.
Let’s go
Under Beall’s medical direction, the foundation had spent the 1980s and early 1990s doing things that no other health nonprofit was doing. They’d established relationships with biotech and pharmaceutical companies and catalyzed the creation of drugs to treat CF’s worst symptoms: oral pancreatic enzymes that helped patients digest food and extract nutrients; Pulmozyme to break down asphyxiating mucus; and TOBI to more effectively fight lung infections. The lesson Bob Beall learned from the foundation’s involvement … was that they had the power to fund and direct the development of medicines… Perhaps, Beall mulled, he could use a variation of this strategy to lure a company into creating a cure for cystic fibrosis–from scratch.
In 1994 a biochemist named Roger Tsien and Kevin Kinsella started Aurora Biosciences which aimed to use fluorescence assays to enable massively parallel drug screening for the pharmaceutical industry. Tsien would later win the Nobel Prize in Chemistry for the discovery and development of the green fluorescent protein (GFP). When Beall learned about the service Aurora was offering in 1998 he instantly realized its application for developing a drug to treat CF.
The logic was fairly simple. A GFP-tagged CFTR gene would be inserted into a cell line. But instead of inserting the wild type CFTR gene (i.e. the one that works “normally”), the F508del (or any other CF-causing mutation) would be used. Without any treatment, the GFP-CFTR protein would not reach the cell surface because of its malformed shape. If a drug was able to fix the shape of the CFTR protein then it would reach the cell surface along with the GFP-tag, and the fluorescence would be visible. In other words, if a treatment worked, then the cells should fluoresce. If there was no fluorescent colour, then the protein didn’t make it to the surface, and the treatment didn’t work. Even if the protein reached the cell surface, further tests would be need to confirm whether chloride was moving in and out of the cell. While many of the specific technical details would need to be worked out, the principle made sense.
Aurora’s technology meant that a plate of thousands of wells could be constructed, each containing cells with the mutated protein, and a different drug could be put into each of wells. Cameras could then check for the intensity of the fluorescence to see which compound worked, and how well. Not only had this high throughput screening never been used to find a drug candidate (up until that point companies had tested a few candidates per day), finding a small molecule that would fix the shape of a broken protein was also virgin territory. However, by end of the 90s, researchers knew that many diseases were a result of misfolded proteins including Huntington’s, Creutzfeldt-Jakob, Gaucher’s, and others. From Aurora’s perspective, if it “could find a molecule to refold this protein, maybe it would serve as a road map for treaiting other devastating conditions.”
But screening molecules was going to be expensive. Where would the CFF get the money for this? In June 1998, the CFF’s nonprofit discovery arm sold the rights of the royalties of TOBI back to PathoGenesis for $19 million. It was a good start, but they would need more money. As has so often been the case in the history of CF, it was the parents of children with CF that proved the decisive element. Paul Flessner was a senior executive in Microsoft and had specifically moved to Seattle due to the quality of their CF care center for his child. Flessner knew that the CFF was raising money to fund a new round of drug discovery and was able to set up a meeting between Beall, Steve Balmer, and Bill Gates.
The Gates Foundation was convinced, and the CFF received its largest donation in October 1999 of $20 million. By 2000 an agreement had been reached with Aurora. The CFF would pay up to 47 million dollars over the next 8 years, with the same amount of additional funding if the drug went to trial. The Gates Foundation and TOBI royalty money would cover 75% of the needed initial funds.
Beall expected the work to be done within 6-8 years–half the time it took a traditional drug company to make a drug from start to finish… [I]t would also cost only a tenth of what a traditional pharma company would have needed…
The right multiverse
Anyone who knows about the transformative effect that modulator drugs like Trikafta have had for pwCF will find reading chapter 37 of Travedi’s disturbing given how close the whole project was to being shelved. In 2001 a company called Vertex Pharmaceuticals acquired Aurora Biosciences and did a full review of their pipeline after acquisition. Vertex was seriously considering whether to kill the partnership with the CFF. The new owners worried about the opportunity cost of having their scientists focus on a molecule that was unlikely to work, in an indication they had no experience, for a disease with no history of being treated successfully, even if the CFF was footing the R&D bill. Even if a promising compound was found, there would be significant costs associated with organizing clinical trials, setting up the manufacturing process, and developing a molecule that could be taken orally.
Vertex’s CEO and president of research told Beall that they would only be willing to continue the research if the CFF was able to commit to ever increasing sums of money as the drug progressed along the clinical trial path (if it ever got there). Beall was not happy, but realized he had no choice but to agree. The sums of money were going to be big: more than $100 million. For a non-profit like the CFF, sums of money reaching this level were unheard of.
In 2002 the COO of the CFF, Rich Mattingly, was put in charge of raising the $100 million. He knew that only one person could do it: Joe O’Donnell. Joe was a successful businessman and fundraiser who had raised countless money for CF since his son Joey died of cystic fibrosis in the 1970s. Joe was initially uninterested, but Rich wore down Joe’s defenses and he agreed to lead the campaign by 2003. The Milestone to a Cure campaign began in 2004 and would raise over $175 million dollars over the coming decade. These funds would be used to support all of the modulator therapies that would be developed by Vertex.
Vertex also came to appreciate the important morale booster the CF team provided. Many scientists had developed strong emotional connections with the CF community and had met with patients and families. Unlike Aurora, Vertex was actually a pharmaceutical company and knew that they would need to engineer human cells rather than mice cells for the high throughput screening to have any credibility. Vertex developed a “cell core” that produce 20 different cell lines and would eventually use lung cells of CF patients who underwent a transplant to do the screening.
Aurora had developed the technology to be able to carry out experiments on a high throughput scale and measure fluorescence, but they didn’t have any chemists. Joshua Boger founded Vertex in the year that the CFTR gene was discovered. In its early days, Vertex pitched itself a drug-design company, developing molecules which target the underlying 3-dimensional shape of the protein. But Vertex’s early targets weren’t working as well as expected, partly because protein shapes are not stable. Furthermore, knowledge of the structure of CFTR protein was unknown at the time.[19] This was one of the reasons why Vertex acquired Aurora so that an alternative (high throughput screening) method could complement their traditional drug discovery process.
There were two pieces of information that suggested a molecule could fix mutations like F508del. First, early research had shown that when cells which carried mutant CFTR genes, like F508del, when cooled to 30 degrees Celsius, enabled some amount of protein to reach the cell surface (25% of the level compared to the wildtype). Further lowering the temperature could increase yield to as much as 50%. Second, research on a chemotherapy agent called genistein had been shown to change the voltage of cells with the G551D mutation. This was important because the G551D mutation has a CFTR protein that is “appropriately localized to the apical membrane but does not normally conduct”–in other words, it could get to the cell surface but not actually allow chloride ions to pass through.
These two pieces of evidence showed that a pharmaceutical intervention was at least possible in theory. But it also demonstrated that fixing a mutation like F508del might require two or more molecules: one to get the protein to the cell surface (the “corrector”) and one to allow the chloride ions to flow through (“the potentiator”). Using a chemotherapy drug was of course not an option for CF patients, so another drug would need to be found. Vertex decided to dedicate equal resources to finding both the corrector and potentiator compound. Unlike the corrector, if the team could discover a viable potentiator, they would actually be able to develop a treatment for the small percentage of CF patients (5%) who had the G551D mutation.
After more than a year of unsuccessful searching, Vertex made a bold pitch to the CFF: prioritizing the search for the potentiator. While a potentiator-only drug wouldn’t be very valuable for a pharma company like Vertex given the small number of patients with G511D, it would demonstrate that a protein-correcting treatment for F508del was possible. If the potentiator wasn’t possible, then the corrector wouldn’t matter anyways. Plus, a successful treatment would help to build excitement and credibility for the protein-correcting enterprise. Beall and the CFF signed off on the idea.
The focus paid off, and the high throughput screening had yielded a chemical that was showing a promising dose response curve. In order to actually get this chemical powerful enough in concentrations that could be swallowed, the medicinal chemists stepped in. Sabine Hadida was in charge of the next painstaking process whereby the baseline molecule was tweaked, on atom at a time.[20] The CF team was based at Vertex’s San Diego office where Aurora Biosciences was originally based. The distance to the East Coast created something of a parallel world within the company.
By the fall of 2004, the CF team was growing more and more optimistic that they would prevail, even though nearly everyone at Vertex Cambridge, and the industry, still thought that the CF project was outrageous and bound to fail. It wasn’t uncommon for the medicinal chemists at Vertex Cambridge to laugh at the unconventional chemical structures Hadida’s team was producing. The compounds didn’t look the way drug-like molecules usually looked, and the chemistry used to make them wasn’t traditional either. Many of the molecules emerging … violated the Lipinski rule of five–a set of rules chemists used to evaluate whether a drug molecule was likely to be effective. At a lot of pharmaceutical companies, chemists weren’t allowed to make molecules that fell outside that box.
The candidate molecule was called VX-770. The initial tests were stunning. When applied to rat cells with the G551D mutation, the voltage returned to normal levels, and toxicity was not detected. When the CEO of Vertex was told the good news, he suggested the team “visualize it.” The CF team needed to use lung epithelial cells if they were going to show how a clogged and sticky cilia could be returned to a normal state. They used an existing F508del cell line (as a G551D line did not exist) and used the temperature trick to get the cells to rise to the surface. VX-770 did its magic, and under the microscope the team was able to record a video of a silent and sticky plate of cells coming to life after treatment. The fact that VX-770 potentiator worked for both a G551D rat cell, and a F508del human lung cell brought to the surface, further demonstrated that the potentiator principle could be separated from the corrector molecule, and that a combination would likely work for a range of CFTR mutations that needed one or both.
After testing the drug in rats, Vertex began planning for a phase I trial in 2006 on a mix of healthy volunteers and CF patients. By 2008, Vertex was moving to phase II of the trial, based on impressive increases in lung function for the small number of CF patients tested earlier. Yet even at this moment, the internal enthusiasm for the CF team’s work was muted at Vertex. The retrospective irony is galling. By 2023, the revenues from modulator therapies for CF would approach $10 billion a year for the company (a number which is expected to grow as more countries sign purchasing agreements). If Vertex had dropped VX-770 and its CF treatments, it would likely have gone out of business several years ago. In 2014 the drug developed from VX-770 would be Vertex’s only source of revenue after its hepatitis C drug Incivek lost almost all of its market share to a more effective competitor, prompting Vertex to lay off 15% of their workforce. Today Vertex is one of the most profitable pharmaceutical companies around protected by a moat of patent protections extended by the modulator therapies’ orphan drug status.
Returning to 2008, the phase II results for VX-770 were stunning. Lung function had increased by more than 10% and sweat chloride levels had dropped by an average of 30 to 50 points, often times below the threshold required for a CF diagnosis (60 mmol/L). With these results, the companies top executives finally began to take the CF team’s work seriously.
But for Van Goor, the Phase II data was more than just a sign of a promising medicine. It was one of the most important pieces of data in the history of cystic fibrosis. There was Paul Quinton’s discovery of the malfunctioning chloride channel. There was the discovery of the CF gene. And now there was this: the discovery that fixing the CFTR’s broken channel with a synthetic molecule could change a patient’s entire physiology, from the saltiness of their skin to the volume of air in their lungs.
A distinction with a difference
In 2008, as the US and world economy crashed under the weight of toxic mortgage-backed financial securities, the world of CF drug development would accelerate. Vertex was able to move VX-770 to a phase III trial rapidly due to the funding and operational commitments that the CFF signed up to. Since almost every pwCF is treated at a CF care center and has been genotyped, the CFF’s clinical trial network was able to identify and contact almost every person with G551D and recruit the needed patient sizes that would be required for a phase III trial to demonstrate efficacy. By 2011 the phase III trial was completed and showed equally large and statistically significant effects to what were augured in the smaller phase II trial. The drug was submitted to the FDA with priority review due to CF’s orphan disease status. VX-770 was christened as ivacaftor (trade name Kalydeco), and approved in 2012 to much fanfare.
… it was a huge victory not just for patients and their families, but also for Doris Tulcin and Bob Dresing–both still alive and following the drug trials–who had centralized the foundation, made it solvent, and set out the organization’s mission; for Bob Beall, who had launched university research programs with hefty grants to lure the most talented scientists to this neglected orphan disease and taken a risk investing in a biotech company, and then Vertex, who had created a new type of drug that could fix a broken protein and in the process launched a new era of medicine; and for Joe O’Donnell and his Milestones campaign team, which raised the money that made the new drug possible. It also validated the foundation’s funding model, venture philanthropy, and proved that its targeted approach to drug discovery was possible.
Two problems emerged with the approval of Kalydeco. First, and most importantly, the drug would only work for at most 5% of pwCF, highlighting the need to find corrector molecules that could pair with the potentiator and treat the majority of patients, who had an F508del mutation. Second, Vertex set a list price of almost $300K annually, which revealed that the pricing power for a rare disease drug was going to be substantial. For many in the community, this level of profit violated the implicit social contract of a company which leveraged substantial public and non-profit funds for the development of a life-saving drug. Despite several high-level protestations, Vertex remained firm in its high price for Kalydeco.
Before Kalydeco, the exact CF mutation an individual had tended not to be of much importance,[21] but the modulator paradigm would start to create significant variations and inequalities within a community that had hitherto seemed fairly homogeneous. Since the majority of pwCF are either homozygous (two copies) or heterozygous (one copy) with F508del, a corrector for this mutation plus G511D would enable 90-95% of pwCF to be treated. But not everyone would be so lucky, since some individuals might have two copies of a nonsense mutation like W128X, which has a truncated CFTR gene which could never be corrected since the gene is missing most of its required structure to work properly (i.e. you can’t correct a broken arm if the arm is missing).
[Kalydeco] was the first drug ever developed to treat patients with a specific genetic mutation, and its approval marked the beginning of a new era of medicines developed to match an individual’s genes. It was the beginning of the personalized medicine revolution.
While the G551D mutation was the most prevalent class III CFTR mutation, other mutations like S549R were characterized by a similar defective channel gating and would likely show a similar effect to ivacaftor. Vertex ran a supplementary trial in 2013 with only 39 patients with 10 other rare mutations expected to benefit from the treatment, and was able to expand the label for another 9 mutations that same year with FDA approval. Running supplementary trials at this level of genetic granularity had never been done before and would provide a roadmap for how label expansion could be done for the handful of individuals with rare CF mutations based off of solid clinical trials designed for a more prevalent mutation. From Vertex’s perspective, every 5 patients it could add would bring in more than $1 million of revenue per year.
With more than 2000 CF mutations having been identified, many patients with class III mutations, or mutations in a similar region to G551D, began to ask whether they would benefit from the drug. Juliet Paige had asked her pulmonologist at the John Hopkins CF clinic whether she could try the new drug. She had a heterozygous F508del-G178E mutation combination. Her G178E mutation was ultra-rare, having been recorded in only two other individuals from Italy. Even more frustrating, the Kalydeco label expansion had added G178R to the list of eligible mutations, which was a mutation at the same location in the gene, but led to a different amino acid. Presumably if Kalydeco worked for G178R it would work for G178E?
Even with the high price tag for the drug, Vertex couldn’t run trials for such small patient groups. Of the 2000 mutations, 1700 and 1000 were carried by fewer than 50 and 5 people worldwide, respectively. The head of John Hopkins’ CF clinic at the time, Dr. Michael Boyle (now the President and CEO of the CFF), met with Juliet and wrote to her insurance company to conduct a test. The insurance company agreed to provide a month’s supply of the drug and conduct, in effect, a clinical trial with a single person. Remarkably, but not unexpectedly, her lung function improved by 10 percentage points, and her sweat chloride dropped into the normal range. When she went off the drug, her sweat chloride rebounded and her lung function fell. Returning to the drug once more, the effects were replicated. The insurance company was convinced and agreed to cover the cost of Kalydeco. This n=1 approach for clinical trials was done for about 40 other patients in the US, a completely novel way to expand off-label access, but hardly a scalable solution.
At Vertex, Van Goor published research that showed that rat cell lines developed for specific CFTR missense mutations could be used to identify which mutations would likely benefit from ivacaftor. Since these in vitro tests were reasonably good predictors of response to the mutations tested in the clinical trials, the FDA accepted Vertex’s argument that laboratory evidence could form the foundation for adding new mutations to approved list. In 2017 a further 23 mutations were added to ivacaftor’s indication. This decision marked the first time the FDA had approved a label expansion based on an in vitro assay. The FDA signaled that “in vitro assay data could potentially be used in place of additional small clinical trials when seeking to expand to other population subsets, provided that the drug’s safety profile is good, the disease is well characterized and other criteria are met.”
CF had the advantage that its basic biology was extremely well characterized, and there is a tremendous amount of data measuring CF patients and their genetic and phenotype variations. The problem of how to run a clinical trial for the “rare” CF mutations had basically been solved, as long as clinical trial results worked for an analogous mutation, and a well-calibrated in vitro assay experiment could be carried out.
Money, that’s what I want
In 2007, several years after the VX-770 compound had been found, the screening team at Vertex had finally found their first corrector molecule that had some effect on the F508del cell line when combined with VX-770. This compound, VX-809, was able to raise electrical activity by 14%, which was substantially less than the 50% change seen with VX-770 for G511D cells. Researchers discovered the likely reason: corrector molecules were interacting with the F508del CFTR protein at different stages of folding, making two discrete correctors significantly more powerful than only one. The exact domains of the CFTR protein that needed to be “corrected” were pinpointed by researchers in 2012, “… both ΔF508-NBD1 energetic and the NBD1-MSD2 (membrane spanning domain 2) interface stabilization are required for wild-type-like folding.”
In 2009 however, Vertex wasn’t ready to invest more money in finding a double-corrector compound until the VX-770 and VX-809 combination had shown it could work, at least modestly, in human trials. After ensuring that VX-809 (branded Lumacaftor) was safe in a phase I trial in 2009, the company moved the Kalydeco and Lumacaftor combination to a phase II trial in 2010. This new combination drug, named Orkambi, would be the first therapy to combine a corrector and a potentiator to (partially) fix the F508del’s key folding flaw. The phase II results were, as expected, not as impressive as Kalydeco, since a single corrector had only a limited impact on CFTR production. Furthermore, the new drug combo showed some negative side effects that hadn’t been observed before. Seeing these early results, Vertex and the CFF agreed they would need to invest in a second generation of modular therapies.
Besides concerns about the social contract between Vertex and the CF community, the CFF’s royalty agreement with Vertex would now create a perceived conflict of interest as high prices/profits for modulator therapies would translate into more revenues for the CFF. Would the CFF invest in alternative therapies or companies that might cannibalize its revenue for Kalydeco and future drugs? Furthermore, the year-to-year value of royalty payments was a small fraction of its long-run payouts. Unlike investors who rely on time to improve their rate of return, a mission driven organization CFF has a premium on finding a treatment/cure as soon as possible. Like the antibiotic inhaler TOBI, the CFF would look to sell its royalty stake in the first era of modulators as soon as it could.
In 2014 the CFF began working with the investment bank Morgan Stanley to find a buyer who be willing to buyout the CFF’s rights to the future royalty payments from Kalydeco, Orkambi, and any other modulator drugs that were subject to the agreement the CFF made originally with Aurora Biosciences more than a decade prior. At the time of the sale, it was not clear if Orkambi would be approved given it’s more marginal impact and high price tag. The ultimate buyer was Royalty Pharma which agreed to pay $3.3 billion to the CFF; a sum of money unheard of in the not-for-profit space.
Unsurprisingly, not everyone was happy with the agreement, and many thought it would diminish future donations by giving the CFF the appearance of a private equity company rather than a disease-focused charity. Others argued that the money should go to help patients who could not afford Kalydeco, and still others thought it was a darker sign that the CFF has implicitly accepted the sky-high prices that would be charged for future treatments.
Despite concerns from some segments of the community, the CFF stuck to their principle that this money was simply to be reinvested into the next generation of research. In this sense, the money was well timed. The CFF had identified strategic parallel therapeutic strategies including a new model of gene therapy (CRISPR was named by Science in 2015 as the breakthrough of the year) and a new class of double-correctors that might bring Kalydeco-like benefits to more than 90% of the CF population.
A new era begins
Vertex and the CFF had continued the search for more corrector molecules which could be chemically optimized to produce the largest effect possible for CFTR protein expression and chloride conductance. By 2018 Vertex had started phase II trials for two different triple combination therapies. The potentiator VX-770 was paired with VX-661 (an improved version of VX-809) and either VX-445 or VX-659 (second generation correctors). Like Kalydeco, the trial results looked extremely promising and the same combos moved to phase III the next year. When the trial results were released, they caused a sensation:
VX-445 had emerged as the best corrector. The triple that included this talented molecule could even raise the FEV1 of patients with one F508del mutation and one minimal-function mutation–one of the most difficult gene pairs to treat–by a colossal 14.3 percent, and could drop sweat chloride by 45 points. Combined, these three molecules… resurrected the broken CFTR protein. The correctors tweaked the protein at two separate sensitive stages, refolding it and enabling a substantial quantity of it to reach the surface of the cell. There, the doorman helped the channel open, allowing chloride to flow in and out just as it would in a person without the disease, and restoring the balance of salt and water in the lung, gut, pancreas and sweat glands.
Vertex submitted this winning combo, now called Trikafta, for priority review. At the end of 2019, the FDA approved the drug to anyone who had at least one F508del mutation and was over the age of 12. A year later, the FDA expanded the indication to include another 177 rare mutations using the same in vitro assay approach that had been done with Kalydeco before. The term theratyping was coined to describe the process of matching “therapies, or medications, to specific types of mutations.” The year after that, patients as young as 6 could now also take the drug.
More than 90% of CF patients in the US were now eligible for a drug that could return cellular function to near normal levels. As mentioned at the beginning of this review, some studies now suggest that the life expectancy of a person born with Trikafta-eligible mutations would exceed 71 years of age, a number close to the population average of the United States (77).
It’s not uncommon to hear Trikafta referred to as a “miracle drug”, a term which is seldom earned in an industry known for proclaiming every therapy a “breakthrough.” While the randomized control data shows the average change in measured clinical endpoints, only by hearing the personal anecdotes of individuals starting Trikafta for the first time, can you begin to get a sense of how life with CF has changed completely:
That warm morning in March is etched into my memory. While my husband and I sat on the bed reading instructions for how to take Trikafta, it felt surreal. I wanted to manage my expectations, but I remember my stomach was flipping. The excitement, anticipation, and hope were overwhelming. Soon after that, my friend, Mary, called me on FaceTime. We had agreed that my first dose of Trikafta would be taken together, and we did just that – and celebrated with our glass of milk in hand while both of our husbands cheered us on. After we hung up, I cried more tears of hope and happiness as my husband hugged me.
It was a few days before I experienced what CFers called “the purge”. For the first time in my life, my lungs and sinuses had thinner mucous that I could cough up. I remember being both amazed and grossed out by what came out of my body. And I remember whispering to my lungs one morning, “You’re doing it! Keep it up!”
As time passed, we quickly realized that not only would Trikafta stabilize me, but it would also increase my lung function and appetite. I was eating all the time and I was putting on much needed weight. Not only that, but I was no longer out of breath when I ate or when I walked from our bedroom to the living room.
Seeing my life change before my eyes with Trikafta was a humbling and emotional experience. Whenever I took my two yellow morning pills, I would often think of friends who weren’t able to take it or had passed away before this became available. I remember being overwhelmed with gratitude and thinking how privileged I am to not only have access to this medication, but to be able to take it without severe complications or side effects.
It took Bijal Travedi 8 years to write this book. She did hundreds of hours of interviews and must have spoken to countless individuals. One of the most beautiful and charming things in this narrative history is the reference to what must be only a fraction of the amazing stories of pwCF who have contributed to medical advances we see today. When describing the clinical trial of Pulmozyme, we learn about Isabel and Anabel Stenzel, two identical twins with CF who were studying at Stanford when they heard about the trial in 1992. Anabel’s lung function changed only modestly whilst Isabel’s shot up. They knew that the drug worked and that Anabel had received the placebo. These sisters were almost a living embodiment of a potential outcomes model in statistics.
Like Kalydeco, the price tag for Trikafta is around $300K USD. Access within the US can therefore require substantial bureaucratic hurdles depending on the insurance provider an individual has, and many US individuals without the right coverage or ability to meet co-pays have to apply for Vertex’s patient assistance program.
The international picture is even more uncertain for pwCF. In Canada for example, all provinces cover the drug as of 2022, but there is variation in the level of lung function required to qualify, and treating physicians need to file special forms. Furthermore, Canada’s national regulator (Health Canada), does not recognize the in vitro assay approach the FDA accepted to expand Trikafta’s indication to 177 rare mutations, meaning that some Canadians do not qualify for Trikafta even though their US counterparts would. In 2023, both Australia and New Zealand decided to cover the drug, with the topic reaching the national debate stage in the latter country. The drug in unavailable in most of the developing world and is being manufactured illegally in Argentina for a fraction of the cost since Argentina’s patent protections for pharmaceuticals are not in accordance with most international trade treaties.
The CFF and CF community more broadly remains committed to finding a complete cure for CF, regardless of the mutation a pwCF is born with. As Rebecca Schroeder in a recent Ted Talk put it:
When nearing the end of a race, you don’t slow down to admire how close you’re getting to the finish line, you step on the gas, you blast through to the end. “Until it’s done” means that we won’t stop until 100% of people with cystic fibrosis have an effective treatment and a cure.
While Vertex effectively has a monopoly on CF treatments, the CFF is actively partnering with other companies, just as it did with Vertex, to develop treatments for the remaining 10% of patients. For example, Pioneering Medicines is developing a “gene writing” system that could correct the DNA at the cellular level, and would work independently of the specific mutation type. Another company, cystic Medicines, is trying to use “molecular prosthetics” to enable functional chloride ion transfer. If any of these alternative approaches work, they would not only provide treatments to those ineligible for modulator therapies, it could also limit Vertex’s pricing power.
A Foundation for All Seasons
In 1996… [m]oney feuds between the chapters and the national headquarters had ceased once the foundation was centralized, restructured, and moved to Washington; all the money raised was sent directly to national to fund critical breakthroughs. The discovery of the gene–though only partly funded by the foundation–had legitimized Dresing and Tulcin’s reboot of the organization and validated Beall’s strategies for accelerating the science. That amplified the community’s trust in them.
How humans are organized and the incentives they face determine almost everything we observe about society. The creation of new economic and scientific products–innovation–is one of the most essential and important activities our species undertakes. Anyone who has worked in an organization claiming to want to innovate knows that actually doing this is not just complicated, it’s a task so difficult that most who try to do it fail.
The CFF is arguably the most successful mission-driven disease-focused organization in the history of the modern world. It has therefore merited several case studies. Travedi’s book shows in great detail how all of the modern CF treatments are unthinkable without the CFF. Cystic fibrosis has raised more money per patient than any other disease in history. The CFF made a series of excellent strategic choices from its founding.
First, the CFF learned from polio that in order to find a cure, you had to be laser focused on prioritizing research above all other competing priorities. The CFF was able to create grants and dedicated research centers by effectively competing for the best scientific minds to work in this disease space.
Second, the CFF understood how to win over friends in high places, and embedded itself in the medical research establishment. The move of its headquarters to Bethesda to be near the NIH and other federal organizations is indicative of this philosophy. Political and cultural power also meant that a rare disease like CF was recognized within the media and as was able to generate national attention through legislation like the creation of National Cystic Fibrosis week.
Third, the CFF and other national CF organizations shaped the clinical treatment for cystic fibrosis. Not only did did every developed country consolidate treatment of CF patients to specialized care centers which enabled a rapid dissemination of treatment knowledge, it created a registry of CF patient data that would eventually enable the rapid trials and personalized genetic treatments that CF patients have today. This helped to lower the barrier to entry for companies wanting to develop therapies in this space since the cost of clinical trials are the largest share of drug development.
Fourth, the CFF pioneered venture philanthropy in the rare disease space and leveraged the tools of American capitalism. The CFF created the Cystic Fibrosis Foundation Therapeutics, Inc. CFFT as its nonprofit drug discovery arm with the goal of structuring drug development contracts. Like a venture capital firm, only a small fraction of the investments the CFF made into for-profit or potentially commercializable research ever paid dividends (literally). The CFF has poured in more than $500 million into venture philanthropy agreements. The CFFT’s general counsel Schaner and Lubitz has “set the market” for venture philanthropy agreements used now by all disease-focused organizations. This includes agreements where payments are linked to specific milestones and interruption license clauses which allow the CFFT to take back ownership of an asset if the company abandons the project. As Kim and Lo put it in their case study
As part of a nonprofit organization, CFFT’s primary goal is to lower the bar for a biopharma company to engage in CF research. When structuring an agreement, CFFT aims to strike a balance that will create an incentive for a company to invest in research, but also, when possible, provide some upside for the CF community if there is a commercial success. Rather than take an equity stake in the company, CFFT takes a royalty interest on any future revenues of a product. This distinction is critical, as CFFT’s interest is in furthering the development of new treatments for CF, not in seeing a specific company succeed or achieving a financial return. In some cases, the return is capped. The trade-off is limited control on the part of the CFF during drug development, and no ability to set prices or to influence the biopharma partners’ commercial plans.
From biopharma’s perspective, financing from a disease-focused nonprofit provides two advantages: significant clinical expertise in the disease, and a low cost of generally non-dilutive capital. The capital provided by a disease-based nonprofit like the CFF is often less encumbering than traditional venture capital or that provided by a large pharmaceutical company, allowing biotech companies to pursue programs with higher risk but higher reward than they might otherwise be able to sustain.
However, the CFF also brings extensive non-financial resources and expertise to the drug development process. These resources include scientists on staff to help interpret clinical data and improve study design, the ability to conduct extensive clinical trials quickly and efficiently, and a large network of care centers. Depending on its stake in a particular program, members of the CFF may attend periodic research meetings or participate on scientific advisory committees to provide insight into the disease.
Most important, the CFF accumulates data that can significantly lower the risk of the drug development process, particularly in the early stages where the chances of technical and scientific failure are high. During the development of Kalydeco and Orkambi, the CFF provided access to decades of data from the CF patient registry, which amounted to a natural history of the disease (remarkably, this registry included nearly all U.S. CF patients). Individuals involved in the approval process cite this registry as critical to the speedy FDA approval of Kalydeco.
Other nonprofit disease groups for Duchenne muscular dystrophy (DMD) and Spinal muscular atrophy (SMA) have also copied the CFF/CFFT model and have impressive results for conditions that are much rarer than CF. For example, CureDuchenne has invested in Sarepta Therapeutics which has developed several gene therapy treatments for DMD and the SMA Foundation was an essential backer in Biogen’s drug Spinraza, which is expected to extend the lifespan of children by several years and improve motor milestones.
Concluding thoughts
The improvements in the care and treatment of pwCF since the first national CF organization was founded in 1955 is a testament to both the power of scientific progress and the ability of organizations to set strategic priorities and achieve real change. In the era of the reproducibility crisis and politicization of science, it feels very easy to be cynical. But Travedi’s book shows that medical and scientific advances can be a force for good. For anyone needing to recover their sense of optimism and sanity, I strongly recommend Breath from Salt. As another review of Bijal’s book puts it:
Though only a lunatic would deem himself fortunate to have a serious disease like cystic fibrosis arise in his family, I read a story like that and think: There is something about cystic fibrosis that summons the best in people, and those who are connected to the story of this disease in any way have had something remarkable thrust upon them – perhaps not a blessing but surely an opportunity that ought not to be squandered.
References
-
Not only has life expectancy increased in CF, but the most recent pharmacologic gains fundamentally “correct” the underlying disease etiology meaning that symptoms and quality of life improve substantially. Historically, individuals with CF had to engage in daily regimens of physiotherapy, nebulizers, and fistfuls of pills in order to stay healthy (as well as taking proactive measures like avoiding social situations for fear of catching infections). ↩
-
As of 2023, there are two main lanes of attacking the disease through pharmaceutical interventions. The first is extending the current small-molecule “modulator therapy” approach that has yielded amazing results to many patients to the 5-10% of CF patients who have mutations that do not seem to work with the current approach. The second is actually solving the problem “once and for all” using gene therapy which would require inserting genetic material into the cells which are not producing the CFTR gene they should be producing (e.g. lung and pancreas). Keep reading the post to learn more about the history of gene therapy with CF and why this time might be different. ↩
-
At the NACFC conference this year, one speaker suggested that CF was becoming like AIDS, a complex disease that causes many comorbidities and requires medical expertise to treat, but with the right care plan can give individuals normal lives. ↩
-
There are varying definitions of a rare disease, but in the US, the NIH and FDA consider a disease to be rare if it impacts fewer than 200K individuals in the country (about 1 in 1650 people). ↩
-
Mutation here can refer to a single point mutation (i.e. one single base pair or “letter” of the DNA being different from what most people have) as well as deletions (most people with CF are missing three side-by-side letters known as the F508del mutation), insertions, and substitutions. ↩
-
CF also causes the skin to taste salty, although this is not a serious health concern in and of itself except for times of extreme heat when the imbalance of salt in the body can lead to dehydration and heat-related illnesses. ↩
-
Basically once the lung tissue becomes sufficiently scarred and damaged then oxygen will never be able to go through that part of the lungs and patients with CF will slowly lose the ability to breathe. ↩
-
Around 1/30 Caucasian American adults are carriers for the disease meaning that if they produce a child with another CF carrier, there is a 25% chance that the child will inherit two copies of the “bad” CFTR gene and develop CF. Although interestingly, the variation in outcomes for children with the same CF mutations can be quite different. This could be because of environment factors (like time of diagnosis and treatment quality) or other complex epistatic factors. ↩
-
I say the “US” since pharmaceutical companies, both US and non-US ones, derive most of their revenue, and likely even more of their profits, from this market. This is partly related to the fact that the US is the largest economy in the world, but even for comparable economies (e.g. the European Union), the US government cannot negotiate drug prices - although this is just beginning to change - so consumers ultimately pay monopoly prices (although technically “payers” like private insurance companies and Medicare/Medicaid pay, they get their money from premiums and tax dollars so the general population pays one way or the other other). There are also all sorts of clever ways the pharmaceutical companies can increase their chance of “winning” at clinical trials (see Malignant) and “ever-greening” their patents, but that is a topic for another day. ↩
-
While it wouldn’t have been known at the time, using sweat chloride concentrations rather than gastrointestinal enzymes would also have detected more pwCF since not all CF variants (i.e. mutations) lead to “pancreatic insufficiency.” ↩
-
If one had to read only a single chapter in Breath from Salt, I would strongly recommend chapter 9 (“Lessons from Polio”), to understand how these crucial insights shaped the coming half-century of CF research and organizational direction. The next most important chapter in the book is chapter 27 Venture Philanthropy–Funding Drug Development. ↩
-
Something that was realized by many during COVID-19 and Operation Warp Speed, but was strangely lost on leading government officials like Fauci and prominent bioethicists. ↩
-
Nasal and lung cells share many similarities: i) they are types of epithelial cells, ii) they both have cilia and mucus production, iii) they both are part of the respiratory system and share similar roles in managing exposure to airborne substances, and iv) they have similar repair and regeneration mechanisms. ↩
-
I estimate it’s 20% as the NIH claims they spend 48 billion on their website, and about 245 billion was spent overall in 2022. ↩
-
Mutations or genes that occur on different chromosomes, or very far apart on the same chromosome, will appear to be statistically independent and observe the “law of independent assortment” that is fundamental principal in Mendelian inheritance. Thomas Hunt Morgan was the first to demonstrate with fruit flies that when looking at two traits (caused by different genes) that were found on the same chromosome, the law of independent assortment no longer held, and that furthermore, the relative deviation of these probabilities could be used to develop a “genetic map” whereby the distance between genes on the same chromosome could be estimated by their relative phenotypic prevalence in generations of offspring. ↩
-
While only a small number of children were born with CF, because 1/30 Caucasians have the mutation, it was assumed that many potential parents would be willing to pay for a newborn screening test for the disease. ↩
-
See A polymorphic DNA marker linked to cystic fibrosis is located on chromosome 7, A closely linked genetic marker for cystic fibrosis, and Localization of cystic fibrosis locus to human chromosome 7cen-q22, all published in the December 1985 edition of Nature. ↩
-
In Paul Offit’s book’s You Bet Your Life he suggests that the success of CAR-T therapies was, in part, due to lessons learned from the Gelsinger tragedy. Specifically, the cytokine swarm that killed Jesse was prevented when Emily Whitehead received CAR-T therapy for her ALL because her physicians knew what to look for based on the failures learned from the first era of gene therapies. ↩
-
The first look at the atomic structure of the CFTR gene was determined in 2016 using the protein equivalent in a zebrafish. ↩
-
Sabine Hadida, along with two other researchers at Vertex, Paul Negulescu & Fredrick Van Goor, were recently awarded the 2024 Breakthrough Prize in Life Sciences for their work on CF modulator therapy development. ↩
-
The exception to this were individuals who had class IV or V CFTR mutations and thus had “partial function” of the CFTR gene. For these individuals, their sweat chloride levels might be in the “indeterminate” range, not high enough to indicate CF but not low enough to indicate normal cell function, but would be expected to have more mild symptoms of CF. As newborn screening become more commonplace, these individuals would be known as CF-SPID, meaning they were screened positive, but had an indeterminate diagnosis. ↩